

抗体融合蛋白(antibody fusion protein)、抗体偶联药物(antibody-drug conjugate,ADC)、双特异性抗体(bispecific antibody,BsAb)、小分子抗体片段、多特异性抗体、抗体-小干扰RNA(siRNA)偶联物(ARC)……,抗体药物在成为“魔弹”的路上从未停滞。
1975年,Kohler和Milstein成功利用杂交瘤技术生产了单克隆抗体(monoclonal antibodies,mAbs)。1986年,全球首个鼠源单克隆抗体药物莫罗莫那单抗(muromonab-CD3)经美国食品与药品监督管理局(US Food and Drug Administration,FDA)批准上市……,抗体药物历经数十年的发展,已经成为生物药中增长最快的领域。
目前,获FDA批准上市的抗体药物已超100个,超80种抗体药物获国家药品监督管理局(National Medical Products Administration,NMPA)批准进口,国内自主研发并成功上市的国产抗体药物近10种。随着更多抗体药物的成功上市,适应症覆盖面也在进一步扩大,涵盖肿瘤、炎症、自身免疫疾病、器官移植排斥、病毒感染等多种疾病。
万物偶联、双抗爆发 治疗性抗体一直在“内卷”
抗体(antibody)又称免疫球蛋白,是由B淋巴细胞接受刺激后产生的糖蛋白,结构呈“Y”字型,具有特异性结合抗原的功能,在人体抵御外界感染性病原体中发挥重要作用。
早期抗体药物是鼠源单克隆抗体,存在免疫原性强,半衰期短等问题。历经数十年的发展,抗体药物从最初的鼠源单抗,逐步发展为人鼠嵌合抗体、人源化抗体及全人源化抗体。通过片段重组、位点修饰、药物偶联等方法,科研人员研发了包括抗体融合蛋白、抗体偶联药物、双特异性抗体、小分子抗体片段等形式多样的抗体药物。
其中,抗体偶联药物(Antibody Drug Conjugate,ADC)是指通过连接子将小分子细胞毒性药物偶联至靶向特异性抗原的单克隆抗体上的一类生物技术药物。其由三部分组成,分别是能够对癌细胞靶向的单克隆抗体、高生物活性的小分子药物以及能够将单抗和小分子药物连接的连接子。
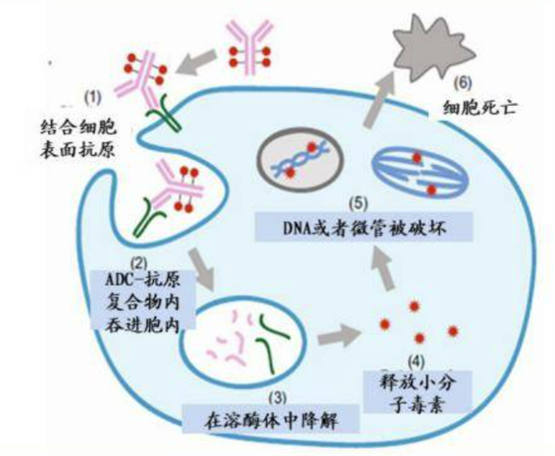
图片来源于网络
当前,随着生物技术和偶联技术的进步,ADC已经突破了传统的抗体+连接子+毒性小分子的模式,多抗偶联药物、核素偶联药物(RDC)、小分子偶联药物(SMDC)、多肽偶联药物(PDC)、双环肽偶联药物、抗体免疫刺激偶联药物(ISAC)、抗体细胞偶联物(ACC)、抗体寡核苷酸偶联物(AOC)等新型偶联药物出现在大众视野。
这些新型偶联药物通过发挥各个偶联组分的优势,提高了疾病的治疗效果,进一步扩大了ADC药物的治疗领域,除了在肿瘤靶向治疗上发挥着重大作用,也拓展到了自身免疫疾病等其他适应症上。
与此同时,双特异性抗体也进入爆发期,将迈向更具临床效果的高成药性平台、联合疗法、多特异性抗体。
据Patsnap新药情报数据库统计,目前全球有800多款双抗在研药物,中国也有400多款在研药物。从全球来看,进入临床阶段的双特异性抗体药物280多款,其中3款申请上市,12款处于III期临床。从国内的研发进展来看,进入临床阶段的双特异性抗体药物接近150款,其中3款申请上市,10款处于III期临床。
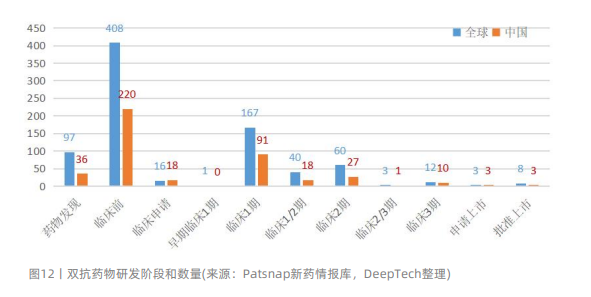
双特异性抗体药物技术获得了极大的进步,多个技术平台的开发解决了不同类型双抗的缺点,如不含Fc片段的双抗(非lgG样双抗)的清除速度快、半衰期较短的缺点,含Fc片段的双抗(lgG样双抗)的重轻链错配问题等。
此外,抗体类型也不限于双特异性抗体药物,已有三特异性抗体(Tri-specific Antibody)、四特异性抗体(Tetra-specific Antibody)等同时靶向多种抗原表位的多特异性抗体(Multi-specific Antibody)药物进入临床研发阶段。
研发成本高、成功率低 治疗性抗体可开发性评估成为重点
抗体药物研发生产技术逐渐成熟与完善,其在生物治疗药物的市场上占据重要地位,但同时其也存在新靶点难以开发、研发成本高、成功率低、临床研究进展缓慢、缺乏规范的标准等问题与挑战。
具体来看,目前主要以人源化抗体研发技术较成熟且成功上市产品占比较大,而以人血浆来源的抗体药物成分复杂,来源稀少,不能满足巨大的市场需求,且存在伦理问题和潜在的危险性,而来源于动物的抗体药物免疫原性较强,容易引起过敏反应,并且病毒及肿瘤的位点突变造成的逃逸,还会导致单位点抗体药物失效。
安全性、有效性等多种原因致使抗体药物的研发成本居高不下、成功率微乎其微。据了解,一个抗体药从临床前到批准,开发费用超过10亿美元。目前全球正在进行I、II期临床试验的抗体药物超过550种,另有79种已经进入开发的最后阶段。但是,即便是人源或人源化抗体,即便已进入到临床试验阶段,最终能够成功开发上市的只有15%左右。
有文献显示,为了降低后期研发失败的风险与损失,提高成功率,临床前阶段就要对候选抗体进行可开发性评估。广义上,可开发性评估包括有效性与安全性、可生产性(manufacturability)、系列理化特性等3个部分。
传统上,开发者用于筛选抗体的首要标准是抗体与相应抗原的亲和力,可用酶联免疫吸附、石英晶体微天平、表面等离子体共振、功能测试等实验进行检测;其次是抗体在动物实验中的药效、药代动力学特征及安全性。
和生产相关的影响因素主要包括抗体生产细胞系稳定性、表达水平、纯化回收率、放大生产性能、制剂稳定性、生产成本等,是涉及细胞工程、抗体工程、发酵工程、药剂学等相关大量实验技术的系统工程。
不少新型抗体药物有效性与安全性挑战较大,研发流程尚不成熟,多处于临床前研究阶段,相关管理规定尚未出台,在一定程度上限制了抗体药物的发展。而研究机构与企业联动合作,充分发挥两者优势,是加速研发有效抗体药物的重要方式。
抗体药物评价超360个 昭衍新药多平台助力其研发上市
作为中国最早从事药物非临床评价的民营CRO企业,昭衍新药具有符合国际规范的质量管理体系(CNAS/ILAC-MRA认证),具备中国NMPA、美国FDA、经合组织OECD、韩国MFDS、日本PMDA的GLP资质以及国际AAALAC和OLAW(动物福利)认证资质,评价资料满足全球药品注册要求。可以向客户提供CDMO、药理药效学研究、生物分析(DMPK)、药物安全性评价研究、临床检测等一站式服务。
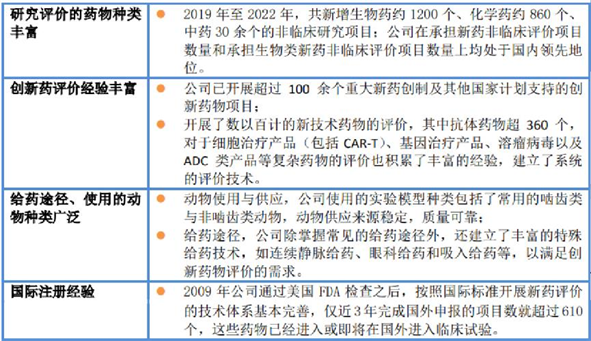
图片来源:昭衍新药2022年度报告
在抗体的非临床评价方面,昭衍新药评价了中国首个获国家药监局批准上市的ADC药物、首个获批用于临床研究的双特异性抗体等。截止2022年,已评价的抗体药物超过360个。昭衍新药针对不同类型的创新抗体产品,提供个性化的设计方案,从药理药效、生物分析方法开发、药代动力学、毒理学评价等方面,不断探索和实践,为新型抗体产品提供全面的高质量的非临床评价服务。
在CDMO方面,2018年成立北京昭衍生物技术有限公司(简称:“昭衍生物”),将以中美两地研发生产基地为依托,为全球创新药研发机构提供可开发性研究、工艺放大优化、质量研究、中试及商业化生产一站式解决方案,是业内唯一能给客户提供“中美两地双厂生产”的服务模式。
昭衍还设有“昭衍药检”,主要面向蛋白药物、疫苗、基因与细胞治疗产品等创新药物质量研究与检定的第三方检测机构,为社会提供创新药物质量标准研究,检定方法建立,标准物质制备及鉴定,细胞库、菌毒种库、原液、成品检验检测,生产工艺质量控制关键步骤如病毒灭活与清除验证等相关服务,致力于支持和促进创新药物的研发及产业化进程。
而为了国内及全球范围内的创新药物品种提供一站式的临床试验样本检测服务,“昭衍临床检测”,具备完善的符合国内和国际GLP、GCP、GCLP,以及CNAS 17025等的质量管理体系,拥有北京、苏州、美国波士顿三处临床分析实验室,专注于大分子药物,特别是抗体药物,基因及细胞治疗药物、小分子药物、生物标志物的临床检测。
当前治疗性抗体领域仍充满挑战与荆棘,作为服务药物创新首屈一指的CRO/CDMO公司,昭衍正在不断地增强自身优势、提高技术壁垒、扩大产能建设、多维度布局科技创新,助力抗体药物的快速研发。
为此,昭衍将分别于2023年5月23日、6月6日、6月20日,晚上19:00-20:30,三期6位老师,系统性的向大家分享新型抗体药物的前沿进展及技术壁垒,共同探讨该领域火热赛道下,新型抗体药物质量研究策略、非临床评价策略、药代动力学特征、临床检测策略等方面内容,欢迎从事本领域的专家学者一同参与讨论!




